The Role Of Nutritional Management In Patients With Classical Homocystinuria In The Saudi Population
Info: 2148 words Sample Topic Selection
Published: 06th Mar 2023
Tagged: Medicine & helathcare
Abstract
Homocystinuria (HCU) is a hereditary metabolic condition characterised by high homocysteine in the blood caused by a cystathionine beta-synthase (CBS) insufficiency caused by a CBS mutated gene. Several bodily systems may be affected by HCU (ocular, central nervous system, cardiac and musculoskeletal systems). The most significant frequency of HCU has been observed in Saudi Arabia, particularly in Qatar, with the kind of pyridoxine non-responsive HCU resulting in severe adverse outcomes. The goal of HCU medication is to maintain homocysteine levels within the body reasonably. According to experts, a low-methionine food should be followed for the rest of one's life. A drug called betaine may indeed assist in decreasing levels of homocysteine. HCU patients should indeed be identified as quickly as feasible. Children must undergo therapy as soon as possible to avoid or reduce the effects of increased homocysteine levels within the body. People with HCU sometimes have a decreased life expectancy if they are not managed. Despite contemporary pharmaceutical treatment, the incidence of HCU in the Qatari population is high. Hence nutritional management is necessary to keep the illness under check. To the best of our knowledge, no prospective research on a limited diet in the Saudi population has been conducted, and we aimed to investigate the dietary outcome. Consequently, we recommended using a planned limited diet to assess the outcomes in HCU patients who visit the hospitals.
Introduction
Classic HCU is an uncommon genetic condition that has a family history. It is due to changes in the CBS genes that prevent homocysteine from becoming adequately broken. As a result, homocysteine and its associated byproducts accumulate in the urinary bladder, muscles, and bloodstream. In childhood, it is related to low IQ, bone loss, musculoskeletal discrepancy, optical lens displacement, cardiac and venous thromboembolism, and psychological disorders (Yan, S., 2022). Worldwide, the prevalence of classical Homocystinuria is 1 in 344,000 (Esser, A. J., 2022). Ireland (1 in 65,000) (Chaudhary, N.,2023) and Saudi Arabia (1 in 1,800) (Mujamammi, A. H. 2022) have more excellent rates of mixed marriages due to higher levels. There are two variants of the classical circumstance: pyridoxine-responsive and pyridoxine-nonresponsive. The physiological response to massive doses of pyridoxine distinguishes responding from non-responsive Homocystinuria. When compared to unresponsive Homocystinuria, B6-responsive Homocystinuria is typically lower. People with Homocystinuria in Saudi Arabia were found to be pyridoxine insensitive, which has serious repercussions (Alhasan, Khalid, 2022). Therapeutic options for CBS insufficiency differ enormously and are determined by the participant's requirements and available treatments. A vitamin B6 supplementation may aid a select people, whereas others might assist most from a low-calorie diet or medication like betaine (Choubisa, D. 2022).
Background
The worldwide experiences regarding prenatal testing for homocystinuria and remethylation abnormalities have been limited. Nonetheless, there is clear evidence that prompt diagnosis by prenatal testing and proper intervention for people with Homocystinuria is beneficial. Moreover, particularly adverse pregnancy vitamin B12 insufficiency, early identification and medication benefit both the infant and the mothers (Gan-Schreier et al., 2010). In theory, newborn screening may identify such illnesses by measuring methionine (Met), the methionine/phenylalanine (Met/Phe) ratio, and total homocysteine (Hcy). The lack of neonatal detection methods often postpones identification. As a result, if individuals are not correctly diagnosed, they face severe protracted death and morbidity. Many people with c.833TNc (p.1278 T) (pyridoxine-responsive HCU) go untreated and may suffer a venous thromboembolism crisis in their third decade (Cruysberg et al., 1996). HCU may now be detected early and treatable with Met-restricted nutrition and betaine, thanks to the establishment a nationwide genetic and biochemical newborn screening (NBS) programme in Qatar in 2006. This has altered the course of the illness over time.
Significance
Nevertheless, current therapy techniques are challenging to sustain, particularly throughout childhood, adolescence, and adulthood, given that they are based on lifelong Met- and nutrient diets containing essential amino acids (Woods et al., 2017). To our knowledge, many dietary procedures have indeed been performed globally and in the Qatari community. However, to the greatest extent of our knowledge, there has been no study of the effects of a limited diet on the Qatari people. Consequently, the suggested study aims to provide HCU-positive children and adults with a balanced diet while also observing the frequency of illness decrease in the Saudi population.
Review of literature
Homocystinuria was initially discovered in 1962 by different organisations in Northern Ireland and the US investigating the amino acid composition of the urine of mentally disabled infants and connected to CBS deficiency in 1964 (Carson & Neill, 1962)—Homocystinuria—the most prevalent sulphur amino acid condition characterized in 1 in 344,000 births. Nevertheless, genetic testing of specific CBS variants in European nations like Denmark, Germany, Norway, and the Czech Republic has yielded estimates between 1:6,500 to 1:20,000. In contrast, biochemical testing of neonates in Saudi Arabia yielded an occurrence of 1:1800. Presbyopia, bone decalcification, and thromboembolisms are among the indications. Location, food, and CBS variation affect their aggressiveness. The condition has 164 CBS mutations (http://cbs.lf1.cuni.cz/mutations.php). The cardiovascular, Brain, ocular, and musculoskeletal systems are most affected by Homocystinuria. The condition could not be evident in newborns. Homocysteine accumulation damages blood artery walls, reducing cerebral blood flow. Newborn screening is best for early homocystinuria detection. Biochemical markers include elevated blood or urinal homocysteine and methionine levels. CBS biallelic dangerous abnormalities may validate genetic screening and identification.
HCU treatment is a low-methionine diet. Betaine therapy lowers blood methionine via several routes. Pyridoxine increases the activity of enzymes in highly bioavailable variants. Pyridoxine nonresponders should consume reduced methionine. A poor diet may treat Homocystinuria if caught early. They include significant psychological disability and incapacity to thrive. The sickness may require daily protein restriction. To reduce homocysteine accumulation, nutrient diets include plenty of methionine. Avoid meat, dairy, nuts, and eggs. Many individuals have been prescribed a protein intake restriction for proper nutrition and homocysteine control. Homocysteine levels help doctors choose the proper treatment for their individuals. Cystathionine synthase (CBS) activities that convert homocysteine to cystathionine decrease. In the following phase of the metabolic process, homocysteine and serine would transform methionine into cystathionine, creating homocysteine. If the CBS is absent or inactive, cystathionine can be generated, and the second pathway is not triggered, raising blood and urine homocysteine levels (Figure 1).
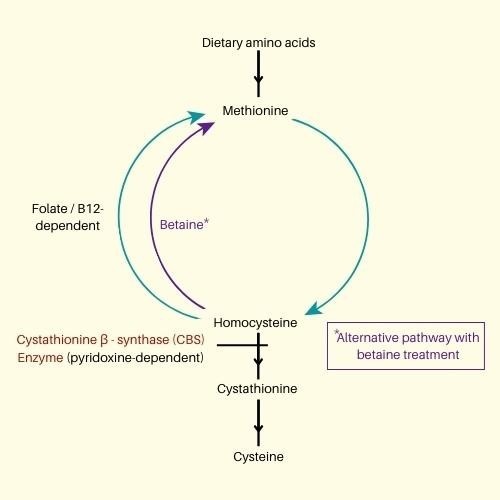
Figure 1. Methionine metabolic pathway
Homocystinuria—the most prevalent sulphur amino acid condition characterized in 1 in 344,000 births (Mudd et al., 2001). Nevertheless, genetic testing of specific CBS variants in European nations like Denmark, Germany, Norway, and the Czech Republic has yielded estimates between 1:6,500 to 1:20,000. In contrast, biochemical testing of neonates in Saudi Arabia yielded an occurrence of 1:1800. Presbyopia, bone decalcification, and thromboembolisms are among the indications. Neurodevelopmental difficulties, inability to thrive and impaired eyesight often contribute to a diagnosis. Other signs are atypical big limbs, chest abnormalities, high foot arches, mental difficulties, and abnormal spine curvature.
HCU treatment is a low-methionine diet. Betaine therapy lowers blood methionine via several routes. Pyridoxine increases the activity of enzymes in highly bioavailable variants. Pyridoxine nonresponders should consume reduced methionine. They are avoiding methionine-rich meals. A poor diet may treat Homocystinuria if caught early. They include significant psychological disability and incapacity to thrive. The sickness may require daily protein restriction. Early identification and preventative and nutritional treatment may stop or cure various illnesses (Sacharow et al., 2017).
Need of the study
Due to cystathionine-synthase (CBS) insufficiency, the CBS mutated gene causes Homocystinuria (OMIM 236200). The origin variation p.R336C is responsible for nearly all CBS deficiencies in Saudi Arabia. The Arab population has severe pyridoxine non-responsive HCU, leading to major clinical complications if not identified and treated early. Individuals who do not get treatment suffer from substantial intellectual disabilities and terrible complex multifactorial illnesses and die sooner. Consequently, the burden of Homocystinuria on the Arab population is significant, prompting the establishment of alternative treatment approaches. Suitable processing choices are restricted and depend on pharmaceutical treatment and life-long methionine-restricted nutrition, which are exceedingly difficult to adhere to (Park et al., 2020). The only option to regulate the HCU of the Saudi population is frequent and meticulous nutrition.
The following are the nutritional modifications for the planned research, following a low-protein diet to decrease methionine intake.
Aim
Saudi Arabia has the greatest CBS insufficiency rate owing to consanguineous marriages and the founding genetic variant c.1006C>T. (p.R336C). Pyridoxine-nonresponsive Classical Homocystinuria has major clinical effects due to such a variant. Pyridoxine-insensitive CBS-deficient HCU treatment relies on a diet. A methionine-restricted nutrition and cysteine supplementation may reduce sulphur inflow and Hcy generation in afflicted persons. Our suggested research will examine these aims.
General objective: Dietary Treatment of Saudi population Pyridoxine Non-Responsive Classical Homocystinuria Patients (Methionine restriction- low natural protein diet with a methionine-free L-amino acid supplement betaine therapy).
Specific objective
I. Assess Homocystinuria before and after the introduction of a limited diet.
Materials and methods
Study design: Hospital-based prospective study.
Study Population: Children and adults with HCU.
Sample size: 200 (100 Children and 100 Adults)
Sampling: Random samples will be collected from the recruited patients visiting the hospital.
Study tool
Questionnaire: After the enrollment of research individuals, data regarding demographic, regular diet, and HCU knowledge are gathered using a well-structured questionnaire method. The surveys will be separated into two groups depending on the planned study's research goals.
Laboratory findings: Blood samples will be collected during each follow-up, and the results will be recorded. Results obtained will be presented prior to the commencement of the dietary plan, and regular follow-up will be provided after the implementation of the restricted diet.
Inclusion and exclusion criteria
Inclusion criteria
Exclusion criteria
Informed consent: Informed consent will be obtained from each participant regarding children from the respective parents. The potential risks and benefits shall be explained to the participants with the maintenance of their confidentiality. All the participants will be asked to fill out the informed consent forms with approval from the local ethics committee for the study.
Facilities available in the institute to carry out the experiments Budget
Work plan: The proposed study is for five years.

Anticipated outcome:
1. Helps in earlier detection by screening for HCU, which eventually aids in minimizing the disease's impact.
2. To minimise possible issues, the identified positive HCU patient would be given a diet plan for illness control, especially in children.
3. Adequate monitoring and follow-up over five years may aid in comprehending the result of the targeted restricted diet.
4. The suggested research might aid in future therapies at various genetic and molecular levels to better understand the condition.
5. dietary planning may be incorporated into the health strategy for further illness reduction effectiveness.
References
Carson, N. A. J., & Neill, D. W. (1962). Metabolic Abnormalities Detected in a Survey of Mentally Backward Individuals in Northern Ireland. Archives of Disease in Childhood, 37(195), 505–513. https://doi.org/10.1136/adc.37.195.505
Cruysberg, J. R. M., Boers, G. H. J., Trijbels, J. M. F., & Deutman, A. F. (1996). Delay in diagnosis of homocystinuria: retrospective study of consecutive patients. BMJ, 313(7064), 1037–1040. https://doi.org/10.1136/bmj.313.7064.1037
Gan-Schreier, H., Kebbewar, M., Fang-Hoffmann, J., Wilrich, J., Abdoh, G., Ben-Omran, T., Shahbek, N., Bener, A., Al Rifai, H., Al Khal, A. L., Lindner, M., Zschocke, J., & Hoffmann, G. F. (2010). Newborn Population Screening for Classic Homocystinuria by Determination of Total Homocysteine from Guthrie Cards. The Journal of Pediatrics, 156(3), 427–432. https://doi.org/10.1016/j.jpeds.2009.09.054
Mudd, S. H., Levy, H. L., & Kraus, J. P. (2001). Disorders of transsulfuration. In The metabolic and molecular bases of inherited disease. Mc Graw-Hill International Book Co. https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225084718
Park, I., Bublil, E. M., Glavin, F., & Majtan, T. (2020). Interplay of Enzyme Therapy and Dietary Management of Murine Homocystinuria. Nutrients, 12(9), 2895. https://doi.org/10.3390/nu12092895
Sacharow, S. J., Picker, J. D., & Levy, H. L. (2017). Homocystinuria Caused by Cystathionine Beta-Synthase Deficiency. https://www.ncbi.nlm.nih.gov/books/NBK1524/
Woods, E., Dawson, C., Senthil, L., & Geberhiwot, T. (2017). Cerebral venous thrombosis as the first presentation of classical homocystinuria in an adult patient. BMJ Case Reports, bcr2016217477. https://doi.org/10.1136/bcr-2016-217477


